
变化对医疗器械的影响
医疗技术和医疗器械公司对医疗保健行业有着巨大的影响。任何技术的变革都是为了满足世界的需求。与其他公司不同的是,医疗设备公司必须不断地对他们的设备进行更改。
技术或工艺的进步可以为病人提供更好的护理和生活条件。对医疗设备的改变总是被期望是好的,并遵循强制性的监管规范,在这些规范中,安全、提高质量和成本效益的最终目的应该得到满足。

医疗设备的变化可以由多种因素驱动,例如:
- 更新特定的工艺或产品
- 提高产品的质量和安全性
- 因用户/监管机构投诉而作出上市后调整
- 方便产品使用
- 升级技术和更多…
当我们在这里谈论医疗设备的变化时,我们说的是设备的变化或医疗设备的变化。这些变化是根据临界度进行分类的。当涉及医疗设备的设计改变或预期用途的改变时,变更指的是重大意义。
例子
在手术中改变刀的形状以提高其性能。更改软件的预期用途,就好像它只是为了检测问题而设计的,将其更改为解决问题。根据更改对系统的影响,可以将更改分为主要、次要或关键。重大变更是指根据需要在设计、原料或工艺上发生变化。例如,改变原材料或重新设计设备,或改变技术。
- 医疗器械设计与开发是其繁荣的迫切阶段操纵师是为您提供医疗器械设计咨询的指导。188金宝搏网站靠谱吗
次要变更是指不影响产品的变更,例如工作区域的行政变更。关键的变更是当它需要广泛的计划、文档变更和需要大量资金的时候。设计的变化影响形式,适合和功能(FFF)。外形匹配功能与设备的设计有关,并被讨论为“整个装配或组件的物理、功能和性能标准或特征,赋予设备独特的身份,并对变化产生影响。”
在设计变更技术过程中,制造商必须密切关注新的变更或交易的部分是否符合医疗器械的外形匹配特性。在对任何设计或工艺进行任何更改时,制造商应考虑FFF要求。Form定义设备的外观。它是设备的物理属性,即形状,大小,尺寸,颜色或视觉参数是项目的特征。例如,在这里我们可以描述一个间隔器或在末端有一个吸嘴的吸入器塑料管
配合是指一件产品与其他组件相互连接或成为另一件产品的组成部分的能力。例如,我们希望我们的垫片或塑料管正确地适合最终产品Function指的是项目将要执行的动作或功能,因为这是设备的存在。例如,Spacer延长了药物进入肺部的时间,对儿童非常有帮助。
如果我们进行任何更改以提高设备的性能,更改评估团队需要评估提议的更改,并对FFF产生影响。当医疗设备发生了重大变化时,就会出现变化和设计控制管理。
变更和设计控制管理
变更和控制管理的需要是为了满足FDA和其他管理机构的法规要求。这是创造安全产品和满足患者/用户需求的有效途径。这是开发最佳产品的最佳实践。变更和设计控制管理帮助我们学会成功地开发满足客户需求的产品。

创新医疗设备
上述循环很好地描述了开发和交付创新(创新医疗设备)的步骤。FDA和其他监管机构保持系统的方法来控制产品的变化。
变更管理中涉及的步骤
- 确定改变的需要,例如,其中一个案例研究给出了导管标志从一边改变到另一边的例子。
- 改变的理由,如导管标志位置的改变虽然使市场部不高兴,但无关紧要,但分析表明,这可能会影响设备的安全性和有效性。
- 检查更改。这里,它指的是对提议的更改进行技术审查。
- 批准变更
- 记录所有步骤。这是医疗器械改革中最重要的一步
- 将变更通知相关人员
- 要实现更改
- 评估变化
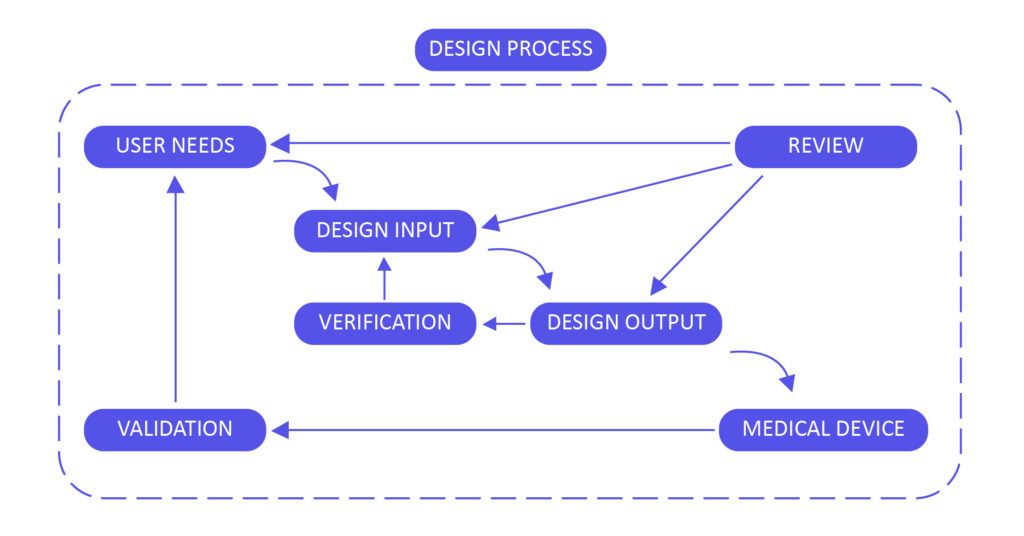
该图描述了设计更改输入和验证的艺术。
改变对医疗设备的影响
在这里,我们将讨论医疗设备在工作或使用方面的主要变化。改变医疗设备是设计改变的重要组成部分,以改善你的产品,使产品获得巨大成功。
医疗设备的变化对成本的发展产生了巨大的影响。生物安全性评估是对医疗器械进行变更时所考虑的参数。这种变化在机器内外都有影响。在此,交流会的角色扩展至影响评估小组,包括技术小组,即研发、品质保证、规管事务、验证、市场营销、工程、品质控制、风险管理及环境安全。
变化影响评估
应在所有可能的领域评估变革的影响,并提供记录和理由。例如,任何对医疗设备的重大改变都肯定会对设备的生物相容性、安全性、有效性和性能产生影响。变更影响评估过程提供了影响决策和所采取行动的记录。
在这里,要考虑以风险为基础的方法,即分析风险相关因素和有益因素。应识别风险相关因素,并指定其严重程度和频率,以确定风险级别并验证其可接受性。变更应集中于产品可用性的效益评估,相关的符合性应最小。为了改善变更的预期目的,需要进行风险评估和管理。测试变化的结果,如材料变化,供应商的变化或材料相似性对医疗设备的变化有重要的影响。请及时更新风险文档。
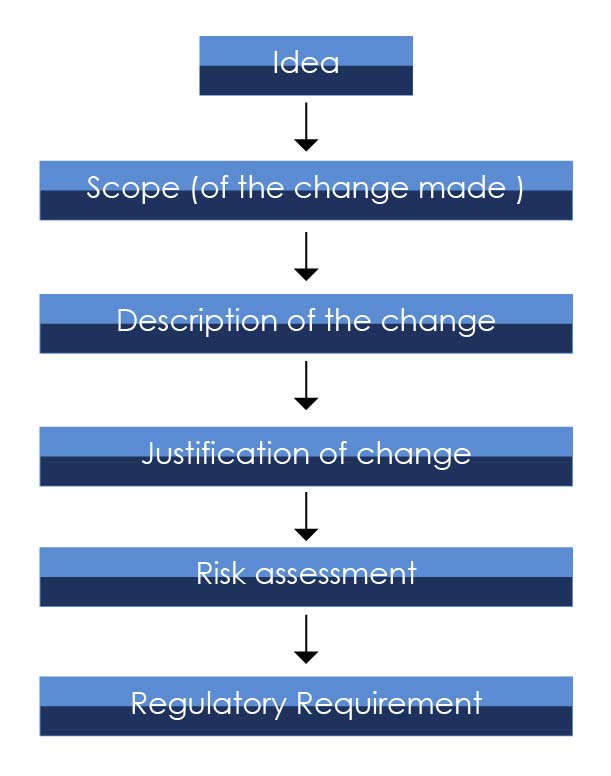
变更评估图包括以下步骤:
监管要求
美国FDA QSR(质量体系法规)(21cfr part 820)和ISO 13485:2016中概述了变更控制的必要性。制造商需要有全面的描述性文档。
- 美国FDA 21 CFR Part 820.30 (i)在描述的地方概述了设计变更
在实施设计变更之前,每个制造商应建立并保持程序,以进行标识、文件化、验证或在适当的情况下验证、评审和批准设计变更。
- FDA 21CFR part 820.40 (b)概述了文件描述的变更
对文件的变更,除另有明确规定外,应由履行原审批职能的个人或组织进行审核和批准。经批准的变更应及时通知有关人员。各制造商应保存文件变更记录。变更记录应包括变更人的描述、受影响文件的识别、批准人的签字、批准日期以及变更生效时的情况。
- FDA CFR第820.70(G)部分概述了生产和工艺变化
(b)生产和工艺改变。每个制造商都应建立和维护规范、方法、过程或程序变更的程序。这些变更在实施前应根据§820.75进行验证或适当时进行验证,这些活动应形成文件。变更应根据§820.40予以批准。
设计和开发变更
许多国家的质量控制标准依赖于ISO标准,ISO 13485:2016制定了一系列的设计控制要求。ISO 13458:2016,医疗器械-质量管理体系-法规要求,第7.3.9节(设计和开发变更的控制)规定:
组织应将控制设计和开发变更的程序编制成文件。组织应确定医疗器械及其预期用途的功能、性能、可用性、安全性和适用法规要求变更的重要性。应确定设计和开发变更。在实施前,变更应为:
- 综述了
- 验证
- 适当的验证,
- 批准
设计和开发变更的评审应包括评估变更对组成部分、过程中或已经交付的产品、风险管理和产品实现过程的输入或输出的影响。变更、变更的评审和任何必要措施的记录应予保持。
欧盟MDR
欧盟医疗器械法规(MDR)为某些医疗器械规定了新的监管义务,如果这些器械的设计或预期用途发生了“重大变化”,它们必须遵守MDR,因此在将其投放欧盟市场之前应该进行相关的MDR符合性评估。
MDGC指导
这为制造商提供了指导,说明对其设备所做的改变是重大改变还是影响了设备的适应症、禁忌症或临床性能。
根据MDR指南,这些变化分为五类
- 目的
- 设计或性能规范
- 原料或材料
- 灭菌或包装设计对灭菌有影响
- 软件
公告机构应评估医疗设备的任何变化及其影响。
结论
改变医疗设备是一种完美的特权,因为我们改变了人们的生活,因为它减少了痛苦,提高了生活质量和责任。新兴技术的安全性是一个挑战,对质量管理的需求是必要的,满足FDA和其他监管要求是必要的,以确保产品的安全性。